Tutorial: Tethered Docking with RxDock
최근에 scaffold-based drug design에 관한 프로젝트를 진행하던 중 scaffold의 위치는 고정해놓고 나머지 구조에 대해서만 도킹을 진행해야 할 일이 생겼습니다. 단백질 pocket에 대한 결합 위치가 알려진 scaffold의 경우 이를 기반하여 디자인된 분자의 도킹된 구조에서의 scaffold의 위치가 크게 바뀌지는 않을 것이라는 생각에 이러한 constraint를 포함할 수 있는 도킹 프로그램을 찾게 되었습니다. 기존에 사용하던 AutoDock Vina 기반의 Smina에서는 구현되어 있지 않은 기능이라, tethered docking이 가능한 도킹 프로그램인 rDock을 사용해보려 하였습니다. 다만, rDock의 많은 기능들에 비해 documentation이 친절하지 않고 한글로 설명 되어있는 도킹 튜토리얼이 없어서 이 포스트를 통해 workflow를 쭉 정리해보고자 합니다.
- RxDock 설치
rDock의 포크 버전인 RxDock을 사용하였습니다. 설치는 간단하게 콘다를 이용하여 설치하였습니다.
conda install rxdock -c bioconda
정상적으로 설치가 완료되었다면 rbdock, rbcavity 등의 rDock의 내장 프로그램들을 사용할 수 있습니다. 사용 가능한 프로그램들은 아래 리스트에서 확인할 수 있습니다.
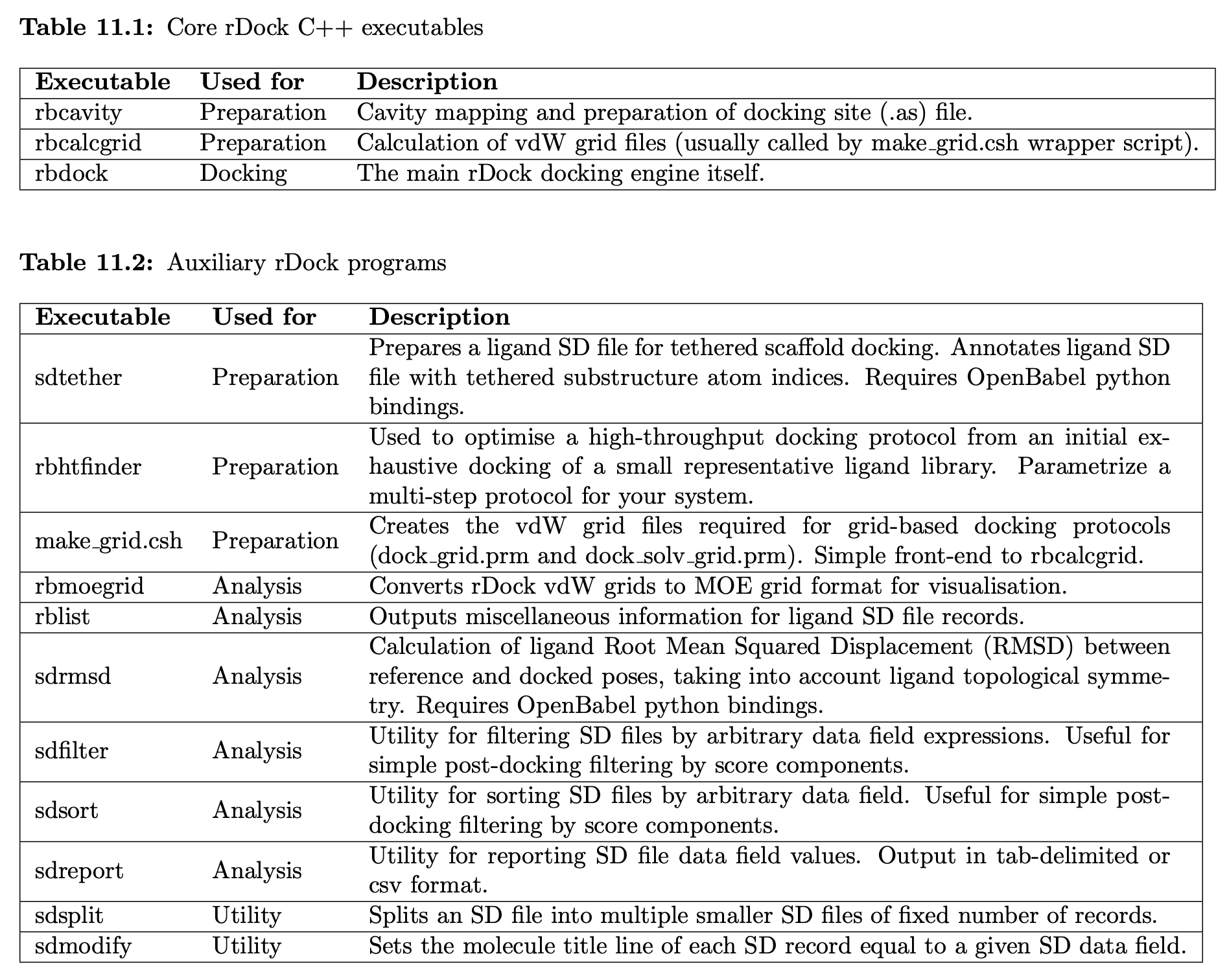
여기서 tethered docking을 위해서 특별히 사용할 기능은 sdtether이며, 다음 섹션에서 중점적으로 알아보겠습니다.
- Ligand 및 Receptor의 Input file 준비
본격적으로 docking을 진행하기 전에 ligand와 receptor에 대해 input 형식을 정리해줘야합니다. (Smina와 다르게 input 파일 형식이 유동적이지 못한 점은 좀 아쉽습니다..) Ligand는 .sdf, receptor는 .mol2로 맞춰주어야 합니다. OpenBabel 같은 파일 변환 프로그램을 이용하여 포멧을 바꿔줍시다.
Tethered docking에 필요한 구조 파일은 총 세 가지입니다.
- Reference ligand (
ref_lig.sdf) - Input ligand, to be docked (
mol_lig.sdf) - Receptor (
receptor.mol2)
추가로, constrain을 줄 substructure를 찾기 위하여 SMARTS descriptor를 필요로 합니다. 아래의 예시에서는 편의상 scaffold의 SMILES를 그대로 사용하였습니다. 이 substructure는 reference ligand와 input ligand에 모두 포함되어있어야 하며, sdtether 프로그램이 reference ligand의 좌표에 기반하여 input ligand의 초기 위치를 결정해주게 됩니다. 또한, mol_lig.sdf 파일의 하단부에 tethered atom의 index들이 찍히게 됩니다. 아래와 같은 command로 해당 프로그램을 사용할 수 있습니다.
sdtether ref_lig.sdf mol_lig.sdf $OUT_LIG_SDF_FILE "$SMARTS"
예시 1) Input command:
sdtether 1jzs_ligand_ref.sdf 1jzs_ligand_0.sdf 1jzs_ligand_input.sdf 'C1COCC(CC2CO2)C1'
예시 2) Standard output:
## Molecule 1 Match Best RMSD reached (match 0, refmatch 0): 4.3253082901238867e-05
DONE
예시 3) Output ligand .sdf file:
1jzs_ligand_0
OpenBabel06142217243D
16 17 0 0 1 0 0 0 0 0999 V2000
-25.6020 6.2160 -29.2220 C 0 0 1 0 0 0 0 0 0 0 0 0
-26.7860 5.6880 -28.3450 C 0 0 0 0 0 2 0 0 0 0 0 0
-27.7850 4.8960 -29.2170 C 0 0 0 0 0 2 0 0 0 0 0 0
-28.3490 5.7690 -30.3511 C 0 0 1 0 0 0 0 0 0 0 0 0
-29.3440 6.8030 -29.7970 C 0 0 0 0 0 2 0 0 0 0 0 0
-30.8100 6.4320 -30.0120 C 0 0 1 0 0 3 0 0 0 0 0 0
-31.3570 5.6130 -28.8280 C 0 0 0 0 0 2 0 0 0 0 0 0
-27.1810 6.4300 -31.1000 C 0 0 0 0 0 2 0 0 0 0 0 0
-26.2300 7.0680 -30.2020 O 0 0 0 0 0 0 0 0 0 0 0 0
-31.7900 6.9960 -29.1050 O 0 0 0 0 0 0 0 0 0 0 0 0
-29.0824 4.8334 -31.3878 C 0 0 0 0 0 1 0 0 0 0 0 0
-24.8181 7.1516 -28.4381 C 0 0 0 0 0 0 0 0 0 0 0 0
-25.1468 8.2895 -27.7553 O 0 0 0 0 0 0 0 0 0 0 0 0
-24.7169 5.0780 -29.8543 C 0 0 0 0 0 1 0 0 0 0 0 0
-24.8181 9.6803 -28.1347 C 0 0 0 0 0 1 0 0 0 0 0 0
-23.5285 6.8735 -28.3623 O 0 0 0 0 0 0 0 0 0 0 0 0
1 14 1 6 0 0 0
1 12 1 0 0 0 0
1 2 1 0 0 0 0
3 2 1 0 0 0 0
4 11 1 6 0 0 0
4 5 1 0 0 0 0
4 3 1 0 0 0 0
6 5 1 1 0 0 0
6 10 1 0 0 0 0
6 7 1 0 0 0 0
8 4 1 0 0 0 0
8 9 1 0 0 0 0
9 1 1 0 0 0 0
10 7 1 0 0 0 0
12 16 2 0 0 0 0
12 13 1 0 0 0 0
15 13 1 0 0 0 0
M END
> <TETHERED ATOMS>
2,1,9,8,4,5,6,7,10,3
- Param file 준비
다음으로 필요한 것은 system에 관한 parameter를 담은 .prm 파일을 만드는 것입니다. rDock에 사용되는 .prm 파일에는 간단한 문법이 존재하는데, 아래와 같습니다.
- 첫 줄은 무조건
RBT_PARAMETER_FILE_V1.00이다. - 아래와 같이
SECTION~END_SECTION문으로 각 섹션의 파라미터를 설정해줄 수 있다.SECTION에는RECEPTOR,LIGAND,SOLVENT,MAPPER,CAVITY,PHARMA등이 있으며 여기에서 각 섹션 별 파라미터의 설명을 찾아볼 수 있다.
SECTION $SECTION_NAME
$PARAM_1 $SETTING_1
$PARAM_2 $SETTING_2
END_SECTION
예시 1) Tethered docking을 위한 system.prm. 이 파일을 통해 SECTION LIGAND의 파라미터를 설정합니다.
RBT_PARAMETER_FILE_V1.00
TITLE test
RECEPTOR_FILE files/1jzs_protein.mol2
SECTION LIGAND
TRANS_MODE TETHERED
ROT_MODE TETHERED
DIHEDRAL_MODE TETHERED
MAX_TRANS 1.0
MAX_ROT 10.0
MAX_DIHEDRAL 10.0
END_SECTION
##################################################################
### CAVITY DEFINITION: REFERENCE LIGAND METHOD
##################################################################
SECTION MAPPER
SITE_MAPPER RbtLigandSiteMapper
REF_MOL files/1jzs_ligand_ref.sdf
RADIUS 6.0
SMALL_SPHERE 1.0
MIN_VOLUME 100
MAX_CAVITIES 1
VOL_INCR 0.0
GRIDSTEP 0.5
END_SECTION
#################################
#CAVITY RESTRAINT PENALTY
#################################
SECTION CAVITY
SCORING_FUNCTION RbtCavityGridSF
WEIGHT 1.0
END_SECTION
Cavity 설정
본격적으로 docking을 돌리기 앞서서 탐색을 진행할 cavity를 설정해주어야 합니다. 이 과정은 reference ligand method 를 통해 진행되며,
rbcavity는 아래와 같은 command로 실행할 수 있습니다.rbcavity -W -d -r $SYSTEM_PARAM_FILEOutput으로
.as파일(docking site)과.grd파일(grid boarder)이 생성되며,pymol로 이를 visualize 할 수 있습니다.예시1)
pymol $GRID_FILE $PROTEIN_FILE $LIGAND_FILE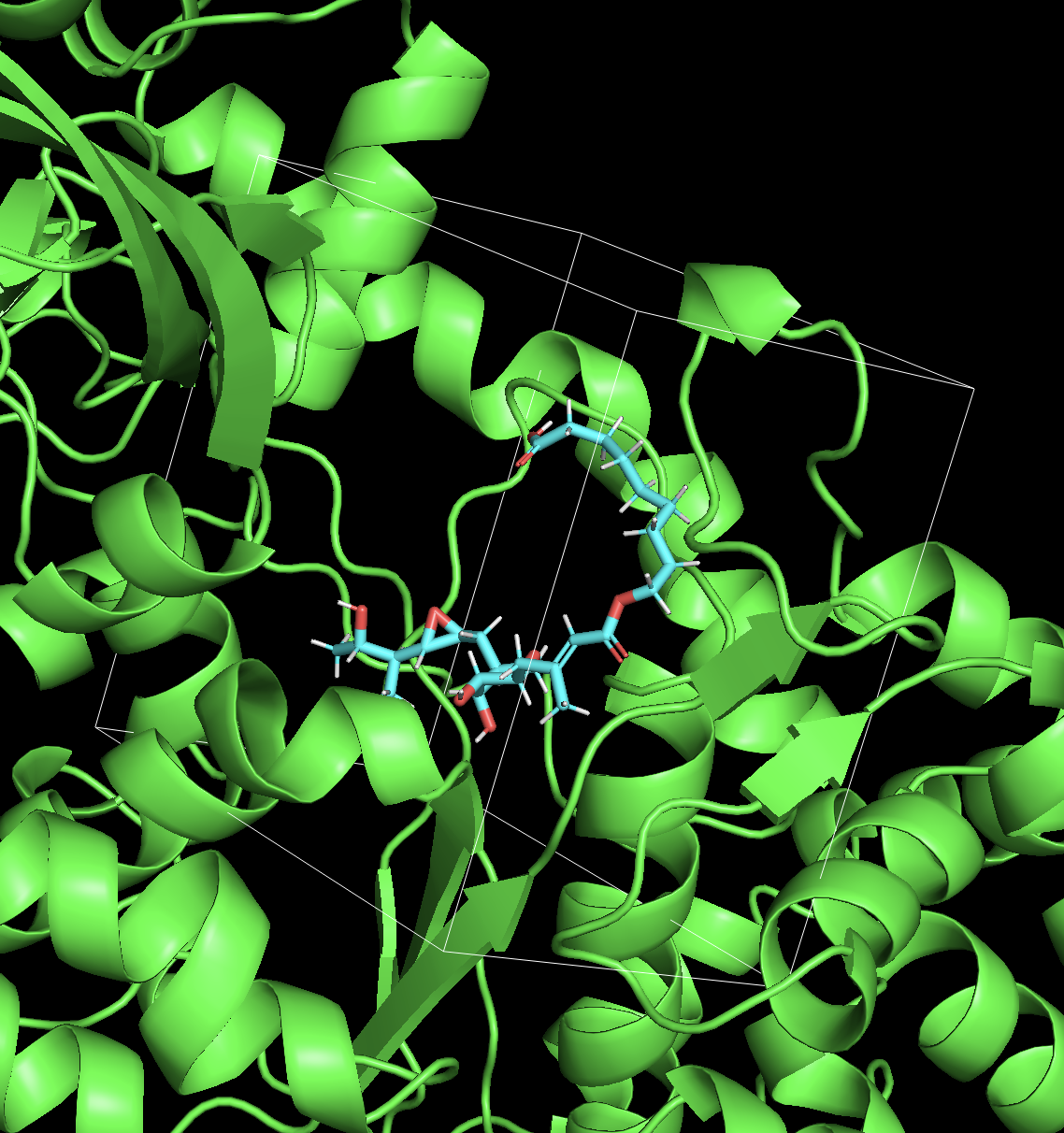
예시2)
isomesh cavity, $GRID_FILE, 0.99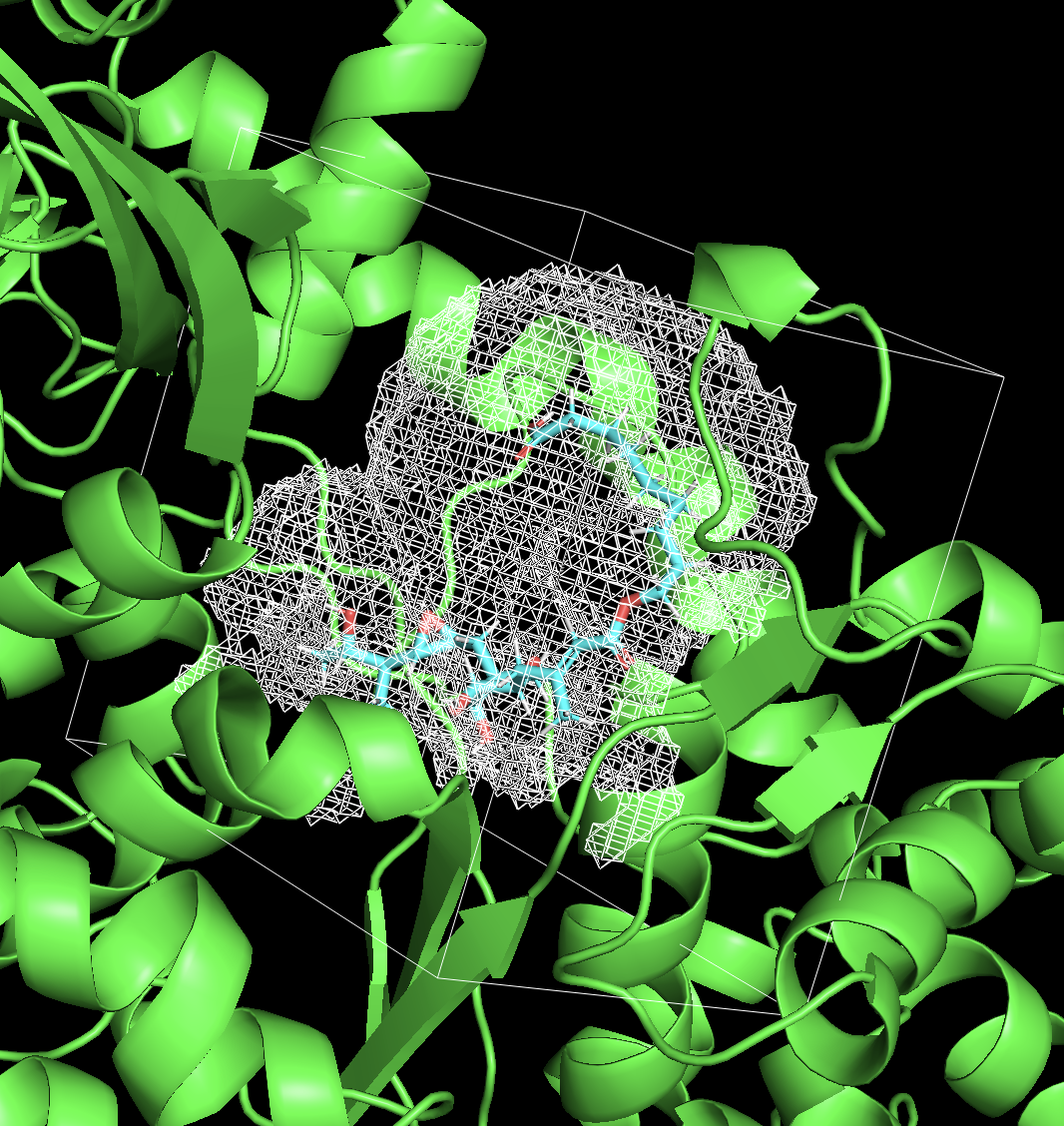
Docking
rDock에서 도킹은
rbdock으로 진행되며, 앞서 준비한 파일들에 추가로 free docking parameter setting을 위한dock.prm이 필요합니다. 이는 여기에서 다운받을 수 있으며, 해당 폴더에 다른 도킹 설정들을 담은.prm파일들도 있습니다.이제 준비는 모두 끝났습니다. 아래와 같은 command로 도킹을 돌려봅시다.
rbdock -i $LIGAND_INPUT_SDF_FILE -o $LIGAND_OUTPUT_PREFIX -r $SYSTEM_PARAM_FILE -p dock.prm -n $NUM_DOCKING_PER_LIGAND --seed $SEED예시1) Tethered docking의 결과
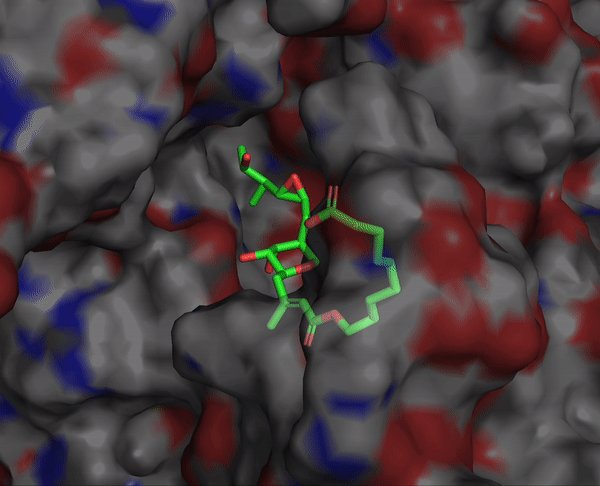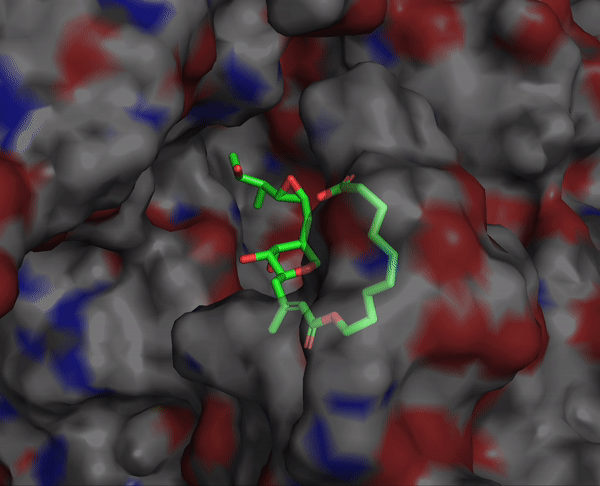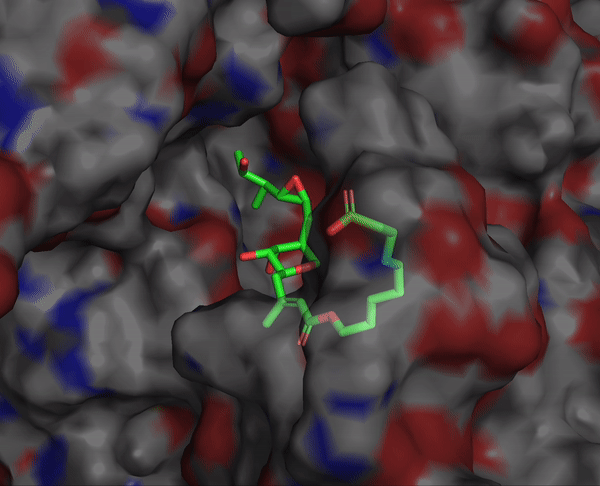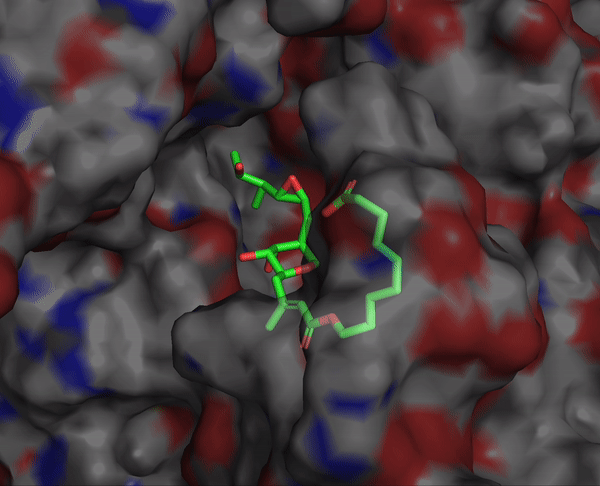
설정한 scaffold의 구조는 거의 바뀌지 않고 도킹이 되었음을 확인할 수 있습니다.
이상 긴 글 읽어주셔서 감사합니다. 🙂
[1] Ruiz-Carmona, Sergio, et al. “rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids.” PLoS computational biology 10.4 (2014): e1003571.
Wonho Zhung
Ph.D. Candidate, KAIST
My research interests include applying deep learning to solve problems in chemistry and biology.